当地时间12月8日,Vertex Pharmaceuticals和CRISPR Therapeutics共同宣布,FDA已批准双方联合开发的CRISPR/Cas9 基因组编辑细胞疗法Casgevy(exa-cel)上市,用于治疗 12 岁及以上患有复发性血管闭塞危象 (VOC) 的镰状细胞病 (SCD) 患者。
上个月,exa-cel获得英国药品和健康产品管理局的有条件上市许可,成为全世界首款获批上市的CRISPR基因编辑疗法。但作为全球公认的标准制定者,FDA批准往往意味着产品拥有了更可靠的背书以及更广阔的市场空间,是CRISPR疗法真正走向商业化的里程碑式起点。
FDA对于CGT等前沿疗法一直在试图寻找有效性与安全性间的平衡点。前不久,FDA针对CAR-T疗法的多起不良事件报告,表示正在调查所有在美上市CAR-T疗法患者中出现恶性肿瘤的情况,并评估监管行动的必要性,在这一领域再掀风浪。这也提醒着药企和Biotech,即使在临床获益显著的情况下,安全性方面的监管也不会放松。
对于CRISPR疗法也是如此,直到10月底,FDA还在召开咨询委员会(AC)会议评估exa-cel基因编辑脱靶的风险。
此次终获FDA批准,无疑是人类技术史的重要一页,也是Vertex强大运营能力下,在罕见病领域和基因治疗领域的新战果,为CRISPR以及基因疗法发展拓开了新的道路。
Vertex和CRISPR如何“后来居上”?
Exa-cel并不是唯一一款针对镰状细胞病的基因疗法。就在同一天,老牌基因治疗公司蓝鸟生物(Bluebird Bio)的Lyfgenia(lovo-cel)也获得了FDA批准,用于治疗12岁及以上患有镰刀型细胞贫血病(SCD)且有血管闭塞事件(VOE)史的患者。
蓝鸟的产品原理是通过慢病毒载体将β珠蛋白基因的功能拷贝递送至患者的血液干细胞中,以产生常规的红血球蛋白。
蓝鸟在2016年时就着手lovo-cel的开发,进入了临床1/2期试验,并于2018年12月获得FDA的突破性疗法认定。2019年7月,蓝鸟生物公司宣布开始一项名为HGB-210研究的临床2/3期试验,以继续评估lovo-cel的安全性和有效性。
对比起来,Vertex和CRISPR公司是后来者。在2013年前后,蓝鸟的核心技术慢病毒载体体外编辑炙手可热,彼时CRISPR公司刚刚成立。2016年,CRISPR公司宣布和Vertex合作开发exa-cel(当时命名为CTX001),但2018年5月,exa-cel针对镰刀型细胞贫血病适应症的临床试验还未开始就被FDA叫停。
不过,Vertex作为有史以来最成功的Biotech之一,应对新药开发上的各种问题可谓轻车熟路。Vertex曾经历经18年开发丙肝药物替拉瑞韦,并在FDA重重施压的听证会上对该药物的副作用与顾问委员会辩论,最终实现全票通过。
历经多次反复讨论和追加数据,5个月后,FDA将exa-cel的临床禁令解除。
CRISPR公司从一开始就拥有了强悍的合作方。之后,exa-cel没有历经太多波折,并于2022年进入临床3期,在当年6月份的欧洲血液病学大会上,Vertex和CRISPR公司公布:7名镰刀型红血球疾病患者在接受exa-cel治疗后长达21个月内不需要输血。
这几年的时间里,exa-cel基本上把创新药研发的认定都拿了一遍。在美国,exa-cel被FDA授予治疗SCD和TDT的再生医学高级疗法(RMAT)、快速通道、孤儿药和罕见儿科疾病的资格。在欧洲,2023年1月EMA和MHRA受理了exa-cel的MAA申请,也授予孤儿药资格以及优先药物(PRIME)资格。在英国,exa-cel还被授予创新护照。
但蓝鸟的lovo-cel没有这么幸运。2020年11月,FDA因CMC原因,将lovo-cel上市申请推迟一整年。次年2月,FDA首次暂停了 lovo-cel 的临床试验,当时有两名入组患者出现疑似血癌,解除禁令仅5个月后,又由于接受治疗的患者出现贫血,FDA在2021年底对lovo-cel临床研究再次进行了部分搁置。

FDA将临床研究置于暂停状态的原因
这极大影响了蓝鸟的进度。按照计划,蓝鸟期望在2023年第一季度末之前向FDA提交BLA,但最终未能实现。反而是Vertex和CRISPR公司在2023年4月4日首先完成滚动BLA申请,蓝鸟则在4月24日才提交BLA申请,第一名的“光环”不再。
抛开公司运作水平不谈,Exa-cel和lovo-cel的竞争,首先是两种技术路线的竞争。慢病毒载体的缺陷是在细胞插入突变时出现致癌基因,目前仍难以解决,而CRISPR技术最多被质疑的是脱靶效应,但可以通过各种策略的优化,例如仔细选择靶位点、优化sgRNA设计和Cas9活性、脱靶检测分析等,以最大限度地降低脱靶效应的安全风险。
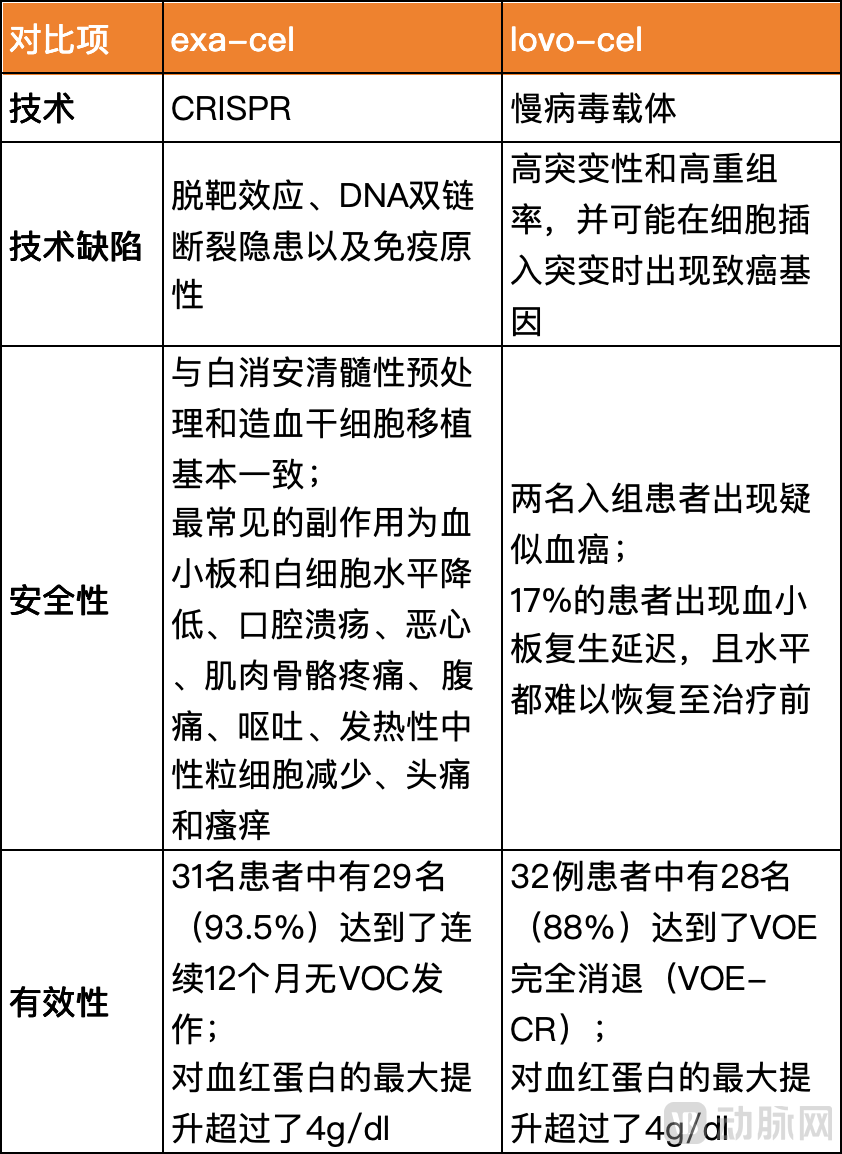
从安全性来看,这展示了新一代技术对已有技术的追碾。蓝鸟基因疗法的研发颇为曲折,尽管已有上市产品,但慢病毒技术不断遭受质疑,CRISPR等基因编辑技术又飞速发展,逐渐占领了大部分的市场注意力和资金。
Vertex能在基因疗法上赚到钱吗?
Vertex作为《十亿美元分子》的主角,以激进的风格、高水平的团队和好运气著称。目前Vertex市值已接近千亿美元,可称为无惧经济周期的创新药龙头。从今年的财报上来看,第三季度业绩指引上调至接近100亿美元,此时exa-cel预期销售额还未计入。
在过往历史中,Vertex的明星丙肝药Incivek和囊性纤维化药物Symdeko、Kalydeco为公司带来了丰厚回报,而这几款药最开始都非Vertex自有管线。Vertex有两大武器:精锐的BD团队,以及能将科学到真正实现商业化的魄力。
在Vertex和CRISPR公司的合作中,Vertex领导exa-cel的全球开发、制造和商业化,双方在研发成本和利润的分配比例最开始是平分,之后修订为Vertex占60%、CRISPR公司占40%。
商业化一直是基因疗法要面对的难题。蓝鸟已有两款上市的基因疗法,但由于价格高昂,商业化并不顺利。为了让支付方减少顾虑,蓝鸟还采取了基于治疗效果付费的方式,即“一次性付全款,治不好可退钱”,但仍未改善公司现金流问题。
对比之下,exa-cel可能拥有的商业化优势包括:
- CRISPR技术在CMC和成本上拥有代际优势。Vertex直接将exa-cel的定价下调至220万美元,低于蓝鸟lovo-cel的310万美元。
- SCD虽然是罕见病,但有较为广泛的患者人群。就像基因疗法的盈利标杆、诺华Zolgensma一样,适应症脊髓性肌萎缩是罕见病中相对常见的一种,患者数量较多。加上exa-cel的疗效和安全性保障,会有一定数量愿意付费的患者。
- Vertex是靠罕见病药物支撑起的公司,对如何在罕见病领域药物上赚钱显然经验丰富,也懂得如何将一条非自研产品带向成功。且其经历过完整的药物生命周期与多个资本市场周期,更何况拥有一般生物制药公司难以企及的战斗力。
- 从Vertex投资来看,它在CRISPR以及整个CGT领域中下了重注,通过收购或联合开发等方式,Vertex在基因编辑技术、递送方式和适应症上多点布局。联想到其创始人博格曾写道:我们不害怕努力、不害怕失败,我们真正害怕的是平庸。以这样的前瞻性与决心,Vertex势必要将CRISPR以及更前沿的基因疗法作为未来的增长曲线。
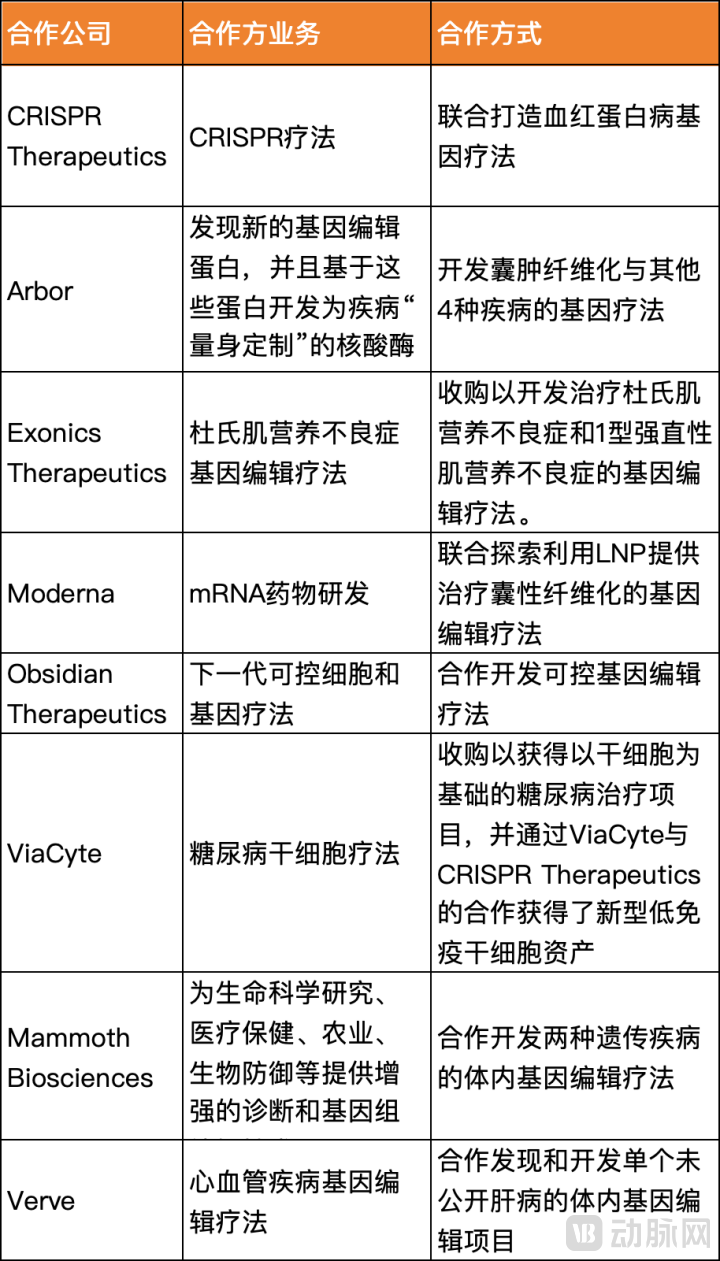
Vertex在基因治疗领域的部分对外合作
CRISPR前路:广泛性和可及性
对于FDA批准exa-cel,有投资人表示:“此次获批的CRISPR疗法仍然是体外编辑,更准确的说,这种个性化治疗应被认为是基因编辑作为工具的某种应用,而再下一代的体内基因编辑才更接近于通用性的‘药物’。”
体内基因编辑覆盖更多适应症、靶细胞和靶器官;周期更优,产品为纳米颗粒或者病毒载体等通用型药物形式,无须个性化的细胞制备流程;最重要的是治疗流程更加可控。上述Vertex的投资版图中也不乏体内基因编辑资产,全球多款处于活跃研发状态中的CRISPR疗法中,有不少是体内编辑项目。
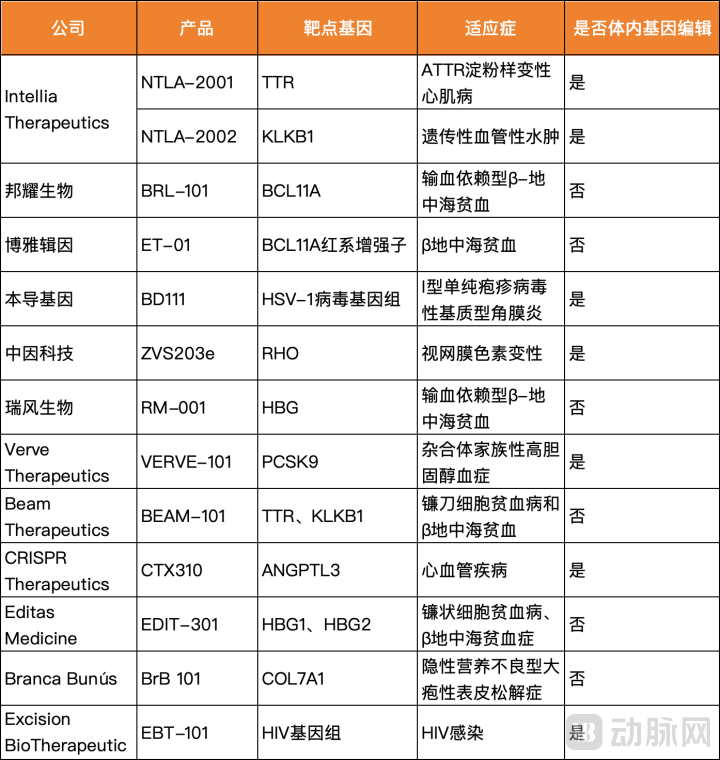
一些业内人士认为,体内基因编辑将是趋势,但目前FDA对于这项技术态度十分小心,Intellia、Verve等体内编辑的先行公司都被FDA出于安全性等考虑叫停。例如,Verve-101这款碱基编辑产品直到上个月才在一年的临床搁置后终于被FDA放行进入美国临床。
不过,在今年Evaluate Vantage发布的基因编辑报告中,多家全球领先的基因治疗公司都表示,体外编辑和体内编辑将各有发展空间。
“体内基因编辑从动物模型到人体应用还存在很多需要解决的问题。不应该孤立地对比体内和体外基因编辑的优劣,也不能简单地认为体内基因编辑的成本一定比体外编辑低。而是需要根据具体疾病和应用场景,从多角度进行评估和选择。”对此,邦耀生物CEO郑彪博士对动脉网表示。
以邦耀生物、瑞风生物等为代表的中国CGT公司,形成了全球基因疗法的另一个高地,这也是十分值得关注的趋势。可以看到CRISPR在研疗法中,中国公司管线占领近半壁江山。
例如邦耀生物针对输血依赖型β-地中海贫血的CRISPR基因编辑治疗产品BRL-101将很快进入临床2期阶段,首个接受BRL-101基因治疗的重型地贫患儿摆脱输血依赖已超3年,这也是全世界首次通过CRISPR基因编辑技术治疗β0/β0型重度地贫的成功案例。代表了国内基因编辑治疗在临床研究方面的突破性成果。
瑞风生物近日公布了输血依赖型β-地贫基因编辑药物RM-001临床Ⅰ期进展,7名IIT患者和9名IND入组临床Ⅰ期患者在治疗后均成功脱离输血依赖,即将开展临床Ⅱ期试验。
本导基因的BD111注射液临床试验申请在7月获美国FDA批准,该药适应症为Ⅰ型单纯疱疹病毒性基质型角膜炎,是全球首个FDA批准的CRISPR抗病毒孤儿药。
瑞风生物CEO梁峻彬博士对动脉网表示:“CRISPR疗法的获批将进一步推动国内基因疗法的研究和发展,尤其是推动监管体系在该药物方向的支撑和完善。基因编辑药物商业化将促使相关部门建立更加完善的指导性文件,确保应用的安全性和有效性,这也有助于规范国内更多其他基因药物的研究。”
中国公司的另一个优势,是可以借助临床和CMC更具性价比的成本上进行创新,从而提高基因疗法的可及性。
郑彪博士介绍:“在国内可以尽量控制各个环节的成本,包括原材料、人工、CMC工艺等,我们在努力实现关键耗材的国产化。例如邦耀生物已与博腾生物达成战略合作,利用其生产优势实现商业化生产。我们也清楚看到了中国医保政策对罕见病领域的支持,相信未来可以让创新疗法惠及更多患者。”
梁峻彬博士也评价道:“基因疗法的成本不仅仅取决于制造成本,还包括研发费用、临床试验成本、监管费用、专利费用等多个因素。整体而言,国内的成本优势是可以预期的,但要推动基因疗法更好的可及性还涉及到医保和商业保险等支付体系以及相关的政策支持、适应症的共识和指南制定、药物的配送能力以及GMP制造的产能等。非常重要的一点是,我们也要给予基因药物等创新性医药产品匹配的价值认可和商业空间,形成良性循环,惠及更多患者,治愈更多疾病。”























