AI领域新突破,腾讯发布首个AI药物研发平台「云深智药」
作者:动脉网
2020-07-09 14:09
7月9日,2020世界人工智能大会云端峰会开幕。会上,腾讯首席运营官任宇昕公布了用AI助力药物研发领域的最新进展——由腾讯自主研发的首个AI驱动的药物发现平台“云深智药(iDrug)”正式对外发布。
云深智药平台的推出,将帮助研发人员提升临床前药物发现的效率,有望缓解新冠疫情威胁下,医药行业亟需快速、低成本地进行药物研发的痛点。腾讯已和多家药企达成合作,将AI模型应用到实际药物研发项目中。目前已有十余个项目,包括对抗新冠病毒药物的相关研发等,在云深智药平台上稳定运行。
“云深智药”的命名出自唐诗《寻隐者不遇》,“只在此山中,云深不知处”,暗含新药研发背后相似的历程。该平台-旨在覆盖临床前新药研发的全流程,包含蛋白质结构预测、虚拟筛选、分子设计/优化、ADMET性质预测(即将开源)及合成路线规划等在内的五大模块。
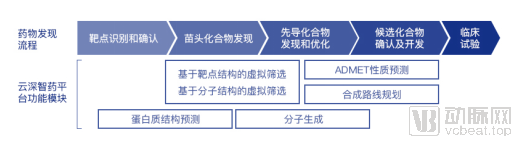
云深智药平台功能模块覆盖药物研发全流程
蛋白质结构预测作为药物设计的基础,对了解生物体内分子间的相互作用至关重要。此前药企、科研机构等通过传统方式进行蛋白质结构的实验测定,往往难度大、周期长、费用高。而通过深度学习模型预测出蛋白质结构以及功能后,计算机可以更快的从数亿的海量小分子中,快速而有针对性地找到潜在的苗头化合物,有效提升研发效率。
此次在云深智药平台上,腾讯AI Lab应用了一项预测蛋白质结构的新算法。数据显示,腾讯新算法在困难案例(hard)上的提高非常显著,比业内公认的权威方法Robetta提高了10%。
自2020年加入蛋白质结构预测的全球权威测试平台CAMEO以来,腾讯AI Lab团队凭借该自研算法,半年内五次夺得月度冠军,领先众多国际知名研究团队,展现了扎实的技术实力。这项算法的创新思路也已应用在云深智药平台上,将在新靶点发现、疾病机理研究上进一步发挥应用价值。
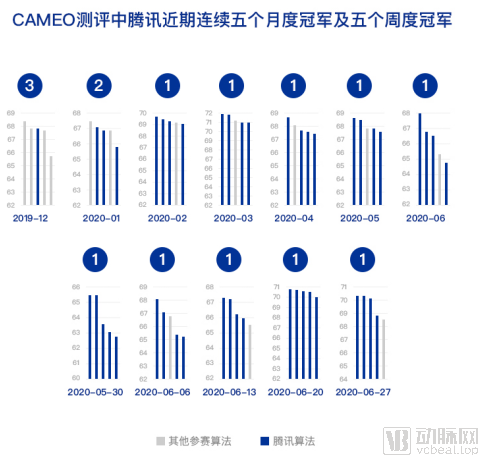
纵轴 lDDT 为蛋白结构预测质量打分,越高表示预测的蛋白模型与真实蛋白结构越相似
在药物虚拟筛选和ADMET性质预测方面,腾讯AI Lab也在多个公开数据集上取得较高精确度、突破了业界标准。后续ADMET预测模块将开源大规模自监督分子图预训练GX模型,分子生成模型预计也将在下半年开源。
目前,虚拟筛选和ADMET性质预测两个工具模块已免费对外开放使用,蛋白质结构预测、分子设计/优化、合成路线规划等模块也将在未来几个月陆续上线,后续平台还将研发更多药物发现功能模块和分析功能。
除了能够免费使用平台搭载的核心功能外,药企、科研机构还可以与腾讯共同开发定制化的AI工具。云深智药平台融合了腾讯AI Lab和腾讯云在前沿算法、优化数据库以及计算资源上的优势,用户不需再自行部署,登录平台就能快速地将AI能力引入现有的研发流程中,可以更便捷地展开研究。
人工智能与大数据作为药物设计领域的关键创新技术之一,将为药物研发带来智能化变革的新机遇。在新基建的背景下,腾讯将继续发力人工智能、大数据等新技术与药物研发需求的深度结合,以先进的技术助力产业、助推我国药物研发行业的高速发展,为医药产业发展创新提供技术支撑。
AI助力药物研发,算法、算力、数据三要素缺一不可、且相辅相成。先进算法可对已有大数据深度挖掘并分析数据间的隐含关系。这个过程不仅直接助力新药发现,还整合了大量已有数据库,同时促进新数据的产生和积累,更好地优化算法。优化的算法反过来也能降低模型对数据量的依赖,提高模型的范化性。腾讯强大的算力支持则加快了数据库存储查找、算法迭代速度,并大大缩短使用模型的运算时间。
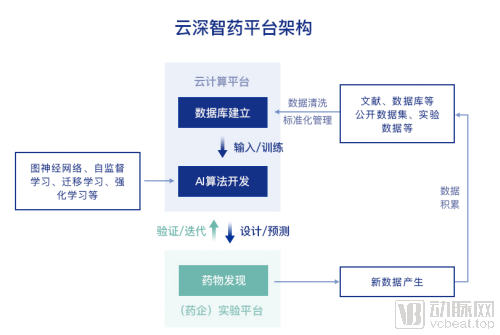
云深智药平台除了在算法领域不断创新,还提供算力和数据库的一体化服务支撑。
数据方面,分子大数据是药物研发中的基础设施。现有的药物分子公开数据集,以PubChem和ChEMBL等为代表,其来源多样。但也由于数据来源于不同机构的不同实验环境,存在数据难以对齐,字段缺失较多,总体质量不佳的问题,从而难以直接用于开发预测模型。云深智药平台使用的分子大数据,基于现有公开数据集,进行了多个环节的精细清洗整理工作,得到可以用于直接构建深度学习模型的药物分子大数据集,并且已在多个药物研发的项目中得到应用验证,清洗过程对多个项目的结果均有很大的提升作用。清洗过后的、打通多个数据库的大数据集已在陆续上线中。
算力方面,腾讯云为云深智药平台提供计算资源,药企、科研机构登录平台即可开展研究,不需要再自行部署,就能快速地将AI能力引入现有的研发流程中。
临床前新药发现流程要经历从靶点的发现和验证、苗头化合物的发现、先导化合物的发现和优化直至临床候选化合物的确认及开发。「云深智药」平台覆盖了临床前新药发现的全流程。
新药发现的第一步是靶点识别和确认,找到药物在体内的作用位点,确定靶点蛋白质的结构是其中的关键工作,被视为药物研发的重要基石。比如一个蛋白参与了某个疾病并成为关键通路上的重要一环,那么当研究人员了解该蛋白的结构后,就可以针对性地设计药物分子来调节蛋白的功能。实验测定蛋白质结构往往难度大、周期长、费用高;通过深度学习模型预测出蛋白质结构以及功能后,计算机便可以更快地从数亿的海量小分子中,快速而有针对性地找到潜在的苗头化合物。
「云深智药」平台采用的蛋白质结构预测方法在准确度上达到国际领先水平,得益于两项关键技术上取得突破。一是基于自监督学习的蛋白质折叠方法,不依赖同源序列,而是直接从序列数据库中通过自监督学习,学得共进化的模式,从而能够从无到有地产生出含有共进化信息的伪同源序列,并最终让这些蛋白能够有效折叠;二是通过一种基于深度学习的可迭代方法,有效整合模板建模与自由建模,首次提出了动态的、可迭代的氨基酸对特异性的约束条件,显著提高了建模的精度,从而更好的折叠蛋白。
针对靶点筛选苗头化合物是新药发现的第二步。与传统的实验筛选相比,计算方法进行的虚拟筛选无需消耗化合物样品,能极大节省人力物力。基于配体的药物设计方法(ligand-based drug design,LBDD)是虚拟筛选的常见方法之一,是指从已知的有活性的配体小分子结构出发,学习和建立分子结构与活性之间关系的模型,用来预测新化合物的活性。由于很多靶点的已测得的化合物活性数据非常有限,严重制约了预测模型的准确性。AI方法有望解决这一问题:例如「云深智药」平台的虚拟筛选模块首次将元学习和深度神经网络算法用于LBDD任务,通过AI”迁移“从其他靶点上面学习到的知识(如分子局部结构对靶点结合强度的影响),应用在目标靶点上来提高模型预测精度。目前该算法在数千个实验数据集上预测精度(预测活性与实验测量活性的相关性)的中位数从目前最高记录0.36提升到0.42,且筛选可用模型的百分比从56%提升到60%,突破业界标准。
进入药物研发后期,预测分子的ADMET性质尤为重要(包括药物的吸收、分配、代谢、排泄和毒性)。据统计,因ADMET性质问题引起的药物后期失败的比例高达60%。因此,及早发现并排除成药性欠佳的分子能够大幅降低后期药物研发失败的风险。基于AI的ADMET性质预测能够让药物化学家快速地进行分子结构改造,优化分子理化性质,缩短药物研发的周期,降低实验测试成本。「云深智药」平台的药物小分子ADMET属性预测模块已在多个数据集上比学术界现有最好模型提高3%~11%;在合作伙伴的反馈中,平台的自研算法精度超过现有商业软件6%~37%不等。同时,平台采用了注意力等机制可视化分子中的子结构对结果的影响,提供模型的可解释性。此外,平台还可提供当地版本等灵活的部署形式,保障用户的数据安全。























