专注罕见病领域,Inozyme IPO募资8.95亿元,开发异常矿化疾病新型疗法【海外案例】
作者:陈宣合
2020-08-16 08:00
近日,动脉网获悉,美国生物制药公司Inozyme Pharma在纳斯达克上市(股票代码为“ INZY”),上市当天以每股16.00美元的价格首次公开发行7,000,000股普通股。截至7月30日,行使超额配股权后,Inozyme Pharma总计发行805万股,最终发行规模达1.288亿美元(约合人民币8.95亿元)。
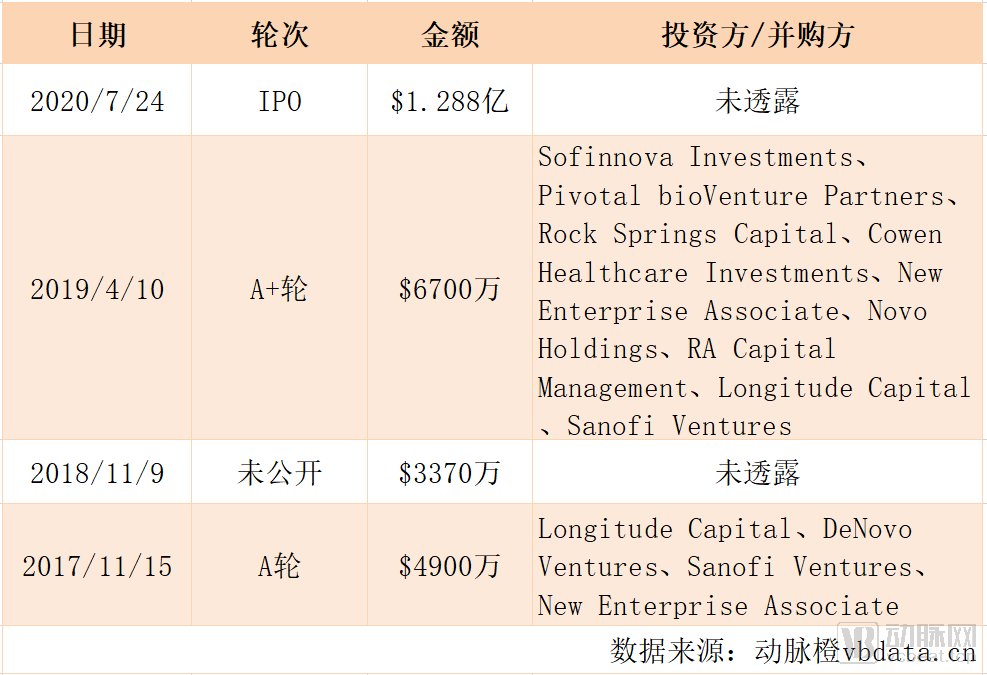
Inozyme Pharma历史融资情况
Inozyme Pharma创立于2016年,总部位于马萨诸塞州波士顿,是一家专注罕见病领域的生物制药公司,致力于开发新型疗法治疗异常矿化疾病,主要包括ENPP1缺乏症、ABCC6缺乏症等。这些疾病会影响人体的血管、软组织和骨骼,进而使人体逐渐衰弱,并可能危及生命。
Axel Bolte是Inozyme的总裁兼首席执行官兼联合创始人,也是IVERIC bio的董事会成员。Bolte本科毕业于瑞士苏黎世的瑞士联邦理工学院,主修生物化学,后在瑞士圣加仑大学进修,获得工商管理硕士学位。
Bolte之前曾在HBM Partners AG(一家生命科学行业的投资顾问服务提供商)中担任风险合伙人、投资顾问,还在Healthcare Venture Capital工作了16年。创办Inozyme之前,Bolte一直专注于医疗保健,尤其是在药品、生物技术、药品开发等方面。他在药品的交易采购、结构设计、联合监控等方面获得了丰富的经验。Bolte还曾是Turos Capital AG的创始人、Allena Pharmaceuticals、Nabriva Therapeutics和PTC Therapeutics的董事会成员。
由于患者相关基因发生病变,导致身体矿化途径代谢异常而引发的异常矿化疾病是一类遗传性罕见病。
在功能正常的矿化途径中,由ENPP1基因编码的蛋白酶将ATP裂解为焦磷酸或PPi,以及单磷酸腺苷或AMP。PPi是有效的矿化调节剂,能控制骨骼中钙晶体沉积的速率。而AMP被进一步代谢为腺苷,腺苷是一种有效的细胞增殖调节剂,在调节血管对损伤的反应方面发挥了很大的作用,同时还负责防止新内膜增生,防止血管内平滑肌细胞过度生长。
ENPP1缺乏症是由ENPP1基因突变引起代谢错误的先天性罕见遗传病,在医学文献中常称其为常染色体隐性低磷酸盐病2型或ARHR2。全世界大概有11,000-12,000个人患有这种疾病。该疾病属于隐性遗传病,由于遗传基因发生突变而导致ENPP1酶活性降低或缺失。
ENPP1缺乏会导致血浆中的焦磷酸或PPi和AMP(腺苷的前体)处于低水平,这会导致血管新内膜增生,伴有较高的早期死亡率和长期发病率。ENPP1缺乏症发生于患者一生中,最早从胎儿发育开始并延续到成年。
ENPP1缺乏的儿童表现特征是血管和器官的持续钙化和佝偻病的发生。由于儿童骨骼动脉持续钙化,促使其骨骼产生一种成纤维细胞生长因子的激素,导致肾脏出现问题,进而患病。该病会导致严重的骨骼畸形,使得患者身材矮小,出现严重骨痛现象,还会增加骨折风险。除此之外,ENPP1缺乏症的儿童可能会因成年牙齿变形而导致关节钙化过多和牙齿问题,使患者的生活质量受到严重影响。
在成年期,除了持续的血管和器官钙化外,ENPP1缺乏症还表现为骨软化症,这会导致严重的骨痛,疲劳,肌无力和骨折的风险。患有ENPP1缺乏症的成年人会出现严重的功能和认知障碍,日常活动能力严重受限,生活质量降低。
ABCC6缺乏症则是由ABCC6基因突变引起代谢错误的先天性罕见遗传病,这种疾病影响了全世界超过67,000个人。ABCC6缺乏症也属于隐性遗传病,由于遗传基因发生突变导致了ABCC6蛋白质活性降低或缺失。ABCC6缺乏也会导致血浆中的PPi水平降低,进而导致全身血管和整个软组织的病理性矿化。ABCC6缺乏症可能会导致失明,使患者产生危及生命的心血管并发症和皮肤钙化。
异常矿化和新内膜增生也可能出现在非遗传疾病中。
脉管系统中的新内膜增生是许多非遗传性疾病的标志,钙化病就是其中之一。钙化抑制是慢性肾脏疾病的一种表现,与低水平的PPi有关,其特征表现为人体皮肤和脂肪中的脉管系统发生病理性钙化现象,进而导致血栓和皮肤溃疡。
这类异常矿化疾病的发病原因虽与基因缺陷类的异常矿化疾病有所不同,但都是由于患者体内的PPi含量不正常所致,这使得Inozyme研发的药物扩大了其适应证范围。
基于这一高度未满足的医疗需求领域,Inozyme开展了INZ-701研究项目。INZ-701是一种可溶性蛋白,旨在纠正因ENPP1和ABCC6基因突变而导致的矿化途径缺陷。上文已经提到,该矿化途径是调节人体体内钙沉积的关键,同时还与新内膜的增生、血管内平滑肌细胞的过度生长有关。
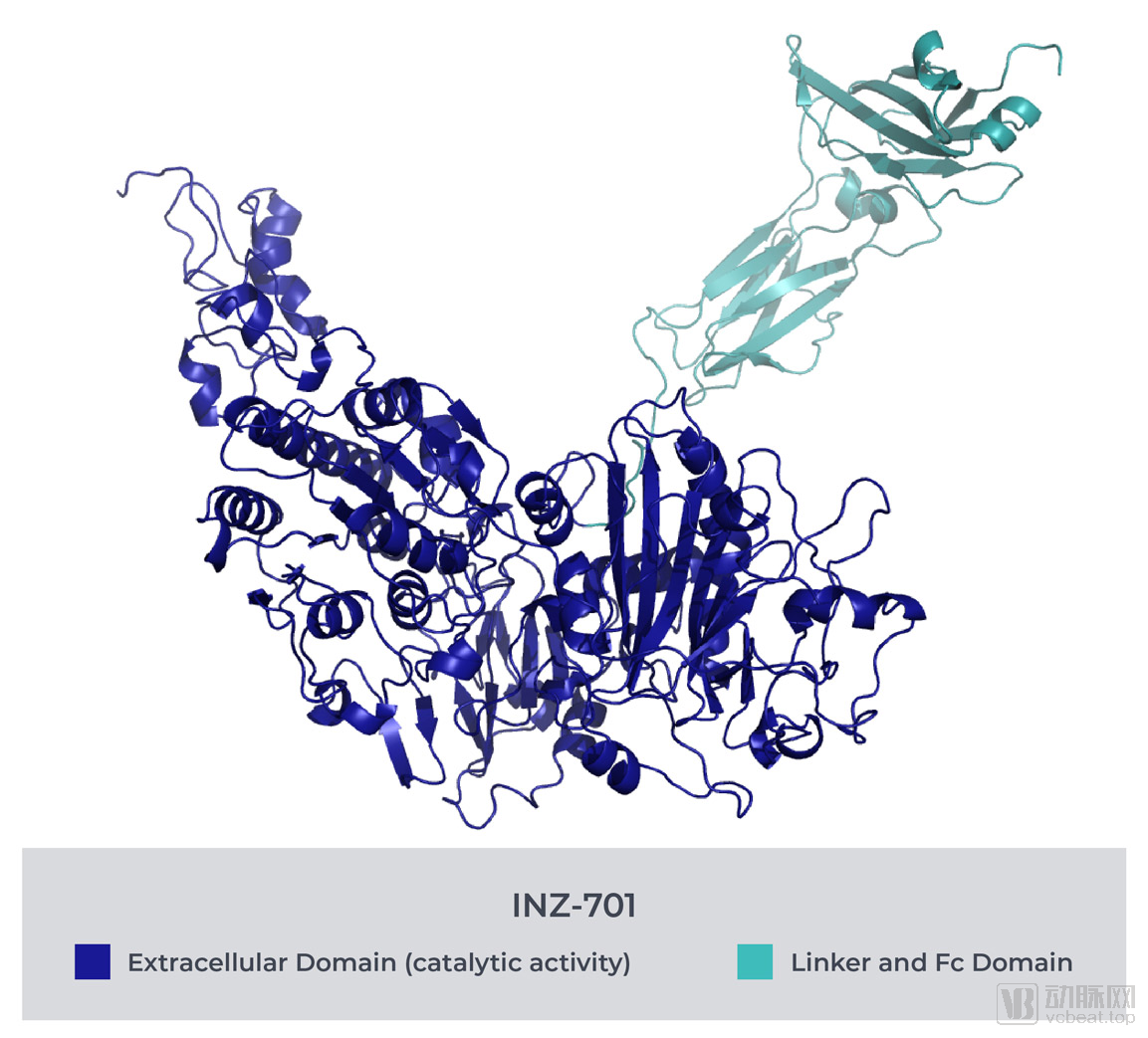
INZ-701通过恢复ENiP1缺乏症的PPi与腺苷的正常平衡含量,弥补因ENPP1或ABCC6单基因遗传病变而丧失的酶功能,以保持人体内环境平衡不受到破坏,使相关代谢过程得以正常进行。
INZ-701的给药方式为皮下给药。给药后,INZ-701会将血液中的血药浓度维持在恒定状态,药效通常也能维持长达一周的时间,解决了频繁间隔时间给药的麻烦。
INZ-701力图“扮演”天然ENPP1的角色。像天然的ENPP1一样,INZ-701催化细胞外ATP和其他核苷酸蛋白进行相关代谢,将ATP裂解为PPi和AMP(腺苷的前体)。
目前,Inozyme的INZ-701项目已经顺利完成了临床前动物实验,有了可靠数据支持后续项目的进行。该开发项目同时也受到专有知识产权的保护。
近年来,为了加大对罕见病药物开发的力度、鼓励制药企业研究罕见病药物,美国政府出台了一系列相关政策。这使得美国一度被很多企业看做是研发孤儿药“重磅炸弹”的摇篮。缩减罕见病药物审批周期、推出研究优先特权等举措让众多药企纷纷加入了开发罕见病药物的洪流。Inozyme无疑是其中之一。
目前,INZ-701已被FDA和欧洲药品管理局(EMA)授予了孤儿药名称,用于治疗ENPP1缺乏症。这意味着INZ-701将拥有50%的税收优惠以及豁免使用费,成功上市后将拥有7年的市场独占权。
Inozyme预计在今年下半年向FDA提交INZ-701的研究性新药申请(IND)和欧洲临床试验申请(CTA),之后会推进INZ-701项目进行两项独立的1/2期临床试验。该实验项目一项在美国和欧洲两个地区进行,专门针对ENPP1缺乏症的患者;另一项在欧洲进行,专门针对ABCC6缺乏症的患者。





















