






2017年5月5日,欧洲联盟(EU)发布了新版医疗器械法规MDR(EU2017/745),以替代旧版医疗器械指令MDD(93/42/EEC)和有源植入医疗器械指令AIMDD(90/385/EEC),同年5月26日生效,过渡期三年。过渡期内,医疗器械厂商可以自主选择以旧指令MDD或新法规MDR申请CE证书;过渡期结束将强制执行新版MDR法规。

欧盟医疗器械法规历史进程节点(动脉网制图)
然而,原计划于2020年5月26日强制执行的新版MDR法规,由于受到疫情等相关因素影响,被宣告推迟一年,直到今年5月26日才正式强制执行。
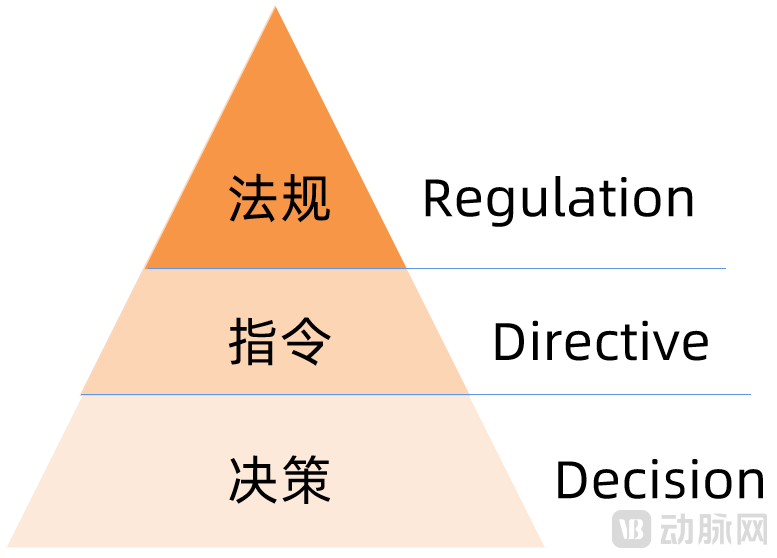
欧盟法规的金字塔体系(约束力由高到低)
从Directive(指令)到Regulation(法规),欧盟提高了对医疗器械的约束力,发布立即在欧盟成员国生效并成为有约束力的法律,此次的Regulation无需像Directive那样需要经过成员国转化成当地法律法规再去落地实施。
值得一提的是,在欧盟医疗器械分类当中,将医疗器械划分为医疗器械(MD)和体外诊断器械(IVD)两大类,目前受到新规MDR管辖的仅限于MedicalDevices,体外诊断器械的相应新规IVDR(EU2017/746)的执行时间为2022年5月26日,也就意味着IVD器械厂商还有一年的时间可以缓冲和适应新规变化,并做好应对准备工作。
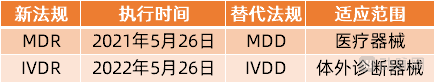
欧盟的两类医疗器械法规及执行时间
对于其他医疗器械(MD)厂商来说,从2021年5月26日开始,如果要申请欧盟认证则必须遵从新规MDR执行。相对于旧法令MDD,欧盟新法规MDR发生了哪些变化?面对这些变化,国产医药器械厂商在申请CE认证时需要注意的点有哪些?对于之前已经申请CE认证的厂商,未来如果继续持有CE证书需要做好哪些应对工作?
在过去,欧盟CE认证的难度较中国NMPA及美国FDA更低,背后的原因则是欧盟医疗器械旧法规的约束力松弛,对医疗器械研发商报批要求普遍较低。同时也导致了部分仅获得CE认证的医疗器械在欧盟地区落地后也常出现医疗事故,所以这些产品未来进入中国及美国市场时,仍然需要面临更长期、更严格的报批流程。
在新规执行之前,一些低风险医疗器械产品研发厂商在申请CE认证时,可以通过自我声明的方式进行申请,但这种申请的方式监管并不严格、缺乏约束力。所以在2020年业内还出现了一则曝光事件,中国深圳某医疗器械公司产品通过自我声明方式出口欧盟,最终发现90%产品不合格。
如果产品没办法精准管控,那么CE认证的“含水量”便较高。据业内人士透露,针对欧盟这种情形,英国、德国甚至还出现过“小名单”——如果你的产品只在欧盟拿了认证,那么该产品在英德申报时还是需要经历严格的临床试验,以确保产品的安全性、有效性。
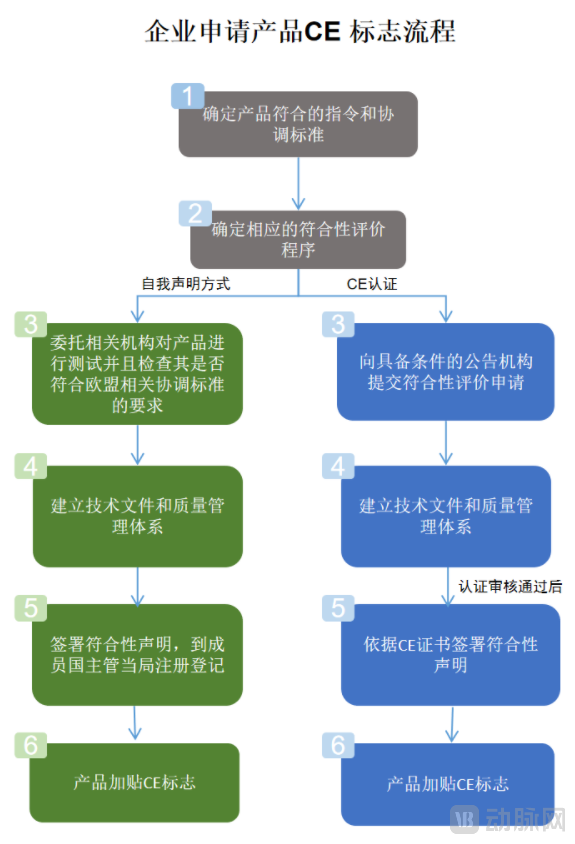
欧盟医疗器械新旧法规申报流程变化(截图自关务小二&中国国际贸易促进委员会官网)
但在欧盟MDR新规执行之后,即便是一些电商平台上进行销售的医用产品也需要通过严格的授权公告机构(Notified Body,NB/欧代)进行申报。与此同时,欧盟对NB机构的要求也大幅提升,公告机构是独立于进行符合性评估活动的产品制造商的第三方机构,要求长期配备具有相关资格证书/资质的产品审查人员、质量管理体系审核人员等,且不能采取外包机制聘用。
对公告机构(NB)的高要求,也让目前获批的能够进行新规公告的机构数量大幅降低。根据欧盟官网信息查询可以看到,过去通过MDD授权的公告机构总计有51家。
然而,自今年5月26日起,这些通过MDD指定的公告机构已不能再根据其指令颁发新证书,而只能对之前颁发的有效证书进行监督过渡。据统计,目前通过欧盟新规MDR授权的公共机构仅有20家。
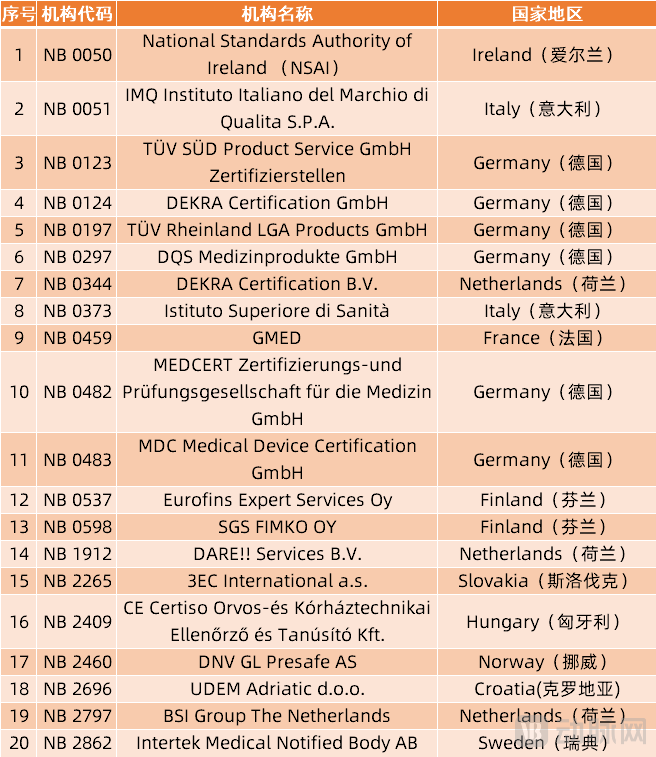
目前能够进行MDR授权公告的20家NB机构(数据来源:欧盟官网)
NB机构数量的骤减,是欧盟认证变难的迹象之一。MDR新规大刀阔斧加强对临床申报的要求,例如在MDR第15条中指出,医疗器械厂商应在其组织架构内至少配备一名负责监管合规的人员,该人员必须在监管事务或与医疗器械相关的质量管理体系方面拥有四年的专业经验;或者拥有如法律、医学、药学、工程或其他相关学科正式资格证明,同时至少拥有一年与医疗器械法规事务或质量管理体系相关的专业经验。
公告机构门槛的提高,认证周期的大幅拉长,新规执行后的欧盟医疗器械CE认证难度将不亚于FDA认证。动脉网(微信号:vcbeat)通过对欧盟新旧法规对比研究,整理出了此次MDR新规变化的几个方向:
1、 医疗器械范围的扩大,以及医疗器械分类的细化。
2、 设立器械唯一标识(UID)和数据库,将强器械溯源管理。
3、 对医疗器械安全性、性能及相关文件的要求提高。
厂商需要在器械上市后建立监控体系,定期更新监控报告。
在欧盟MDR新规中,除了将旧法规MDD管辖范围内的器械纳入医疗器械概念范畴内,还增加了两条特定用途的相关产品也纳入了其中:

欧盟MDR新规“医疗器械”概念扩大(截图自MDR原文)
首先是明确了将基于人体样本进行体外检查(examination)获得信息的相关仪器设备纳入了医疗器械范围内;其次是将专门用于清洁、消毒、灭菌其他医疗器械产品及有源设备的相关器械仪器也纳入了医疗器械范畴。
这里值得一提的是,欧盟将体外诊断(invitrodiagnostic)与体外检查(invitroexamination)做了区分,体外诊断相关器械适用于IVDR(EU2017/746),而体外检查相关器械则适用于MDR(EU2017/745)。
在欧盟法规中,医疗器械被分为4个等级,分别对应Ⅰ类、Ⅱa类、Ⅱb类、Ⅲ类。这是基于人体脆弱性并考虑到与器械技术设计和制造相关的潜在风险为分类原则进行的风险分级。
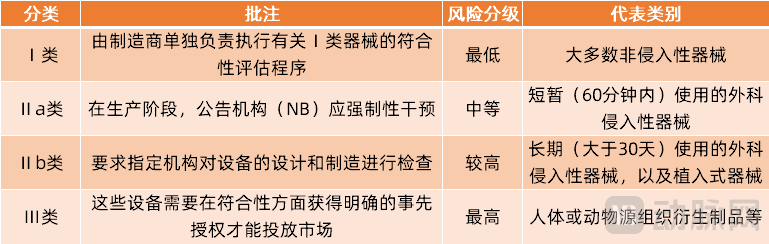
欧盟医疗器械分类
这次欧盟MDR新规中,对医疗器械的基础分类没有变化,但是对具体涉及产品的类目进行了进一步细化,特别是在对Ⅲ类医疗器械上进行了进一步完善。据不完全统计,MDR中增设了12种Ⅲ类医疗器械,它们分别是:
1、 在体外直接接触人体细胞、组织或器官的非侵入性物质组成物。
2、 专门用于与中枢神经系统直接接触的医疗器械。
3、 用于管理医药产品的植入式器械及长期手术侵入性器械。
4、 有源植入式器械或其附件。
5、 乳房植入物或手术网。
6、 全部或部分关节置换物(辅助部件如螺钉、楔子、板等除外)。
7、 椎间盘置换植入物或与脊柱接触的可植入装置(如上,辅助部件除外)。
8、 所有旨在控制、监测或直接影响有源植入式设备性能的有源设备。
9、 提供用于诊断或治疗目的决策软件,其决策可能导致人体死亡或健康状况产生不可逆转的恶化。
10、 具有高等、中等内照射潜力的纳米材料组成的设备。
11、 通过身体孔口引入人体或应用于皮肤并被人体吸收或局部分散的物质及物质组合组成的装置(包括物质及其代谢物被人体吸收达到预期目的/物质及其代谢物在胃或消化道被人体吸收达到预期目的)。
12、 具有集成或合并诊断功能的有源治疗设备,如闭环系统或自动体外除颤器。

MDR相对于MDD分类细化部分
从这次欧盟MDR新规新增的部分我们可以看出,MDR新规在分类上对旧MDD法令进行了进一步细化,对于一些原本模糊的分类进行了边界清晰界定,以及根据同类产品可能带来的不同后果严重程度进行了等级划分。
例如,在对决策软件的分类上,其决策如果可能导致人体死亡或健康状况不可逆转的恶化,该软件则被划分为最严格的Ⅲ类;如果其决策可能导致一个人的健康状况或手术干预严重恶化,该软件则被归类为Ⅱb类;而其余旨在提供用于诊断、治疗、监测生理过程目的的决策软件都被归类为IIa类。
同样,MDR也新增了对包含或由纳米材料组成的设备的分级归类:如果其设备呈现高或中等内照射(internal exposure)潜力,责备认定为Ⅲ类;如果是较低的内照射可能性,则归为Ⅱb类;如果其呈现出的内照射潜力可以可忽略不计,则归为Ⅱa类。
依据欧盟分类的原则,如果该器械或者软件对患者存在更大的潜在影响,都被归类为更严格的管控等级。如MDR法规中第20条分类规则,将除外科手术侵入性器械外,所有与身体孔道相关的、旨在通过吸入给药的侵入性器械均归为Ⅱa类,但是如果该器械作用方式对给药的药物有效性和安全性具有重要影响或它们旨在治疗危及生命的疾病,在这种情况下,则被归类为Ⅱb类。
同时,因为MDR新规重新定义了临床数据的种类和可用性,针对数字健康应用(digital health APP)的医疗器械危险等级分类也会带来重要的区别。目前已经通过德国DVA审核的数字健康应用(DiGA), 需要在基于MDR新规下,确认其产品风险是否升级。如果该APP已经不再满足DVA法案所规定的Ⅰ或 Ⅱa类,升级到Ⅱb类甚至更高危险等级分类,其将不再是DiGA。
除了对医疗器械基础的定义和分类进行了进一步完善,MDR与MDD的还有一大优化在于,欧盟MDR新规要求建立欧洲医疗器械数据库(Eudamed)和对应的唯一器械标识(UID),方便公众充分了解医疗器械相关信息、并实现设备的逐一溯源调查。
医疗器械数据库(Eudamed)需要包含7大电子系统:器械注册电子系统、UDI数据库、经济运营商电子登记系统、认证机构和证书电子系统、临床研究电子系统、警戒和上市后监管电子系统、市场监管电子系统。
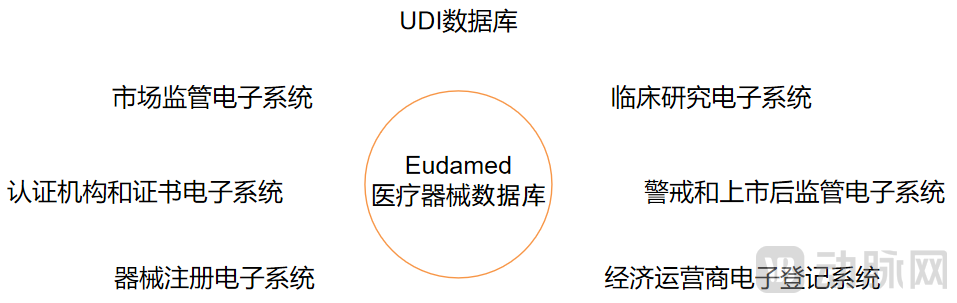
医疗器械数据库的组成系统(动脉网制图)
这七大电子系统的数据由成员国、认证机构、经济运营商和申办方录入,Eudamed进行整理并加工所有信息,最后向认证机构、经济运营商、申办方和公众开放信息访问权限。其中,对于Ⅲ类器械和植入式器械(非定制或研究器械),制造商应起草一份安全和临床性能总结,该总结需要通过Eudamed向公众开放。
这里值得一提的是需要被录入到医疗器械数据库中的UDI数据,UDI数据相当于医疗器械的“身份证”,由UDI-DI和UDI-PI两部分组成。

医疗器械“身份证”UDI组成(动脉网制图)
UDI-DI可以理解为“产品识别码”,由企业识别码和产品规格码两部分组成,是UDI中固定且强制性要求的部分,需要上传到UDI编码数据库向公众公开;而UDI-PI则是器械的“生产识别码”,由医疗器械序列号、生产批号、生产日期、失效日期等生产相关信息组成,非必要性也无需上传到UDI编码数据库。
从全球范围来看,2008年,国际医疗器械监管机构论坛(IMDRF)建立UDI特别工作组,并于2011年9月通过了《医疗器械唯一标识系统指南》;2012年,IMDRF继续补充完善通过了《医疗器械唯一标识系统拟定规则》,指导全球各监管部门依照该拟定规则来构建自己的UDI系统。
目前,在IMDRF的10个国家和地区中,美国和欧盟已经发布了UDI相关法规,不过美国FDA的UDI是由贴标人负责,欧盟的则由制造商负责,且对贴标语言有一定要求,必须是欧盟成员国的官网语言。另外,巴西、中国和韩国也陆续发布了法规的征求意见稿,日本、加拿大和澳大利亚发布了UDI相关通知和指导性文件。医疗器械的全面UID时代即将到来!
在欧盟新规发布之前,依据欧盟旧法MDD报批CE认证难度较低,所以国内不乏一些医疗器械厂商其产品已经在海外通过CE认证,但是在国内却迟迟没有拿下上市许可的情况。
不过此次欧盟MDR新规强制执行之后,将大大提升CE认证的难度,同时提高了CE认证的含金量。在新规中,欧盟强调了提高医疗器械安全性、性能及相关文件要求的重要性,并且加强了器械上市后的监管体系。
在MDR附录Ⅰ第一章一般要求中提出,制造商应建立、实施、记录和维护风险管理体系,其中风险管理应理解为在器械整个生命周期中为连续迭代过程,需定期进行系统更新。并且制造商还需要起草一份包含UDI-DI等8个方面内容在内的安全及临床性能总结给到CE公告机构,并进行验证。后续总结报告还会被上传到Eudamed数据库中接受监管。
此外,MDR中添加了对申报技术文件内容的要求,且明确指出上市后监管计划和安全性更新报告(PSUR)都是技术文件的一部分,并要求依据上市后监管体系收集的资料对技术文件中相应信息进行更新。
医疗器械制造商应计划、建立、记录、实施、维护和更新上市后的监管体系,上市后的监管体系应适于积极和系统地收集、记录并分析器械在其整个生命周期内的质量、性能和安全相关数据,以得出必要的结论,并确定、实施和监测任何预防及纠正措施。
在PSUR上,Ⅱa、Ⅱb和Ⅲ类器械制造商应针对各器械或类别或器械组编制定期安全性更新报告(PSUR),总结数据分析结果和结论,并对采取的任何预防和纠正措施提供理由和说明。其中,Ⅱb和Ⅲ类器械的制造商应至少每年更新PSUR;Ⅱa类器械制造商应在必要时至少每两年更新PSUR。
最后,欧盟新规MDR还丰富了对医疗器械审核的临床评价部分,设立了专门的专家小组以评估审查制造商报批医疗器械的预期临床用途和临床研究方案,制造商应适当考虑专家小组发表的意见,但不得对专家小组就任何未来合格评定程序所表达的观点行使任何权利。
通过分析整个欧盟MDR新规,不难发现欧盟新规的完善一定程度上参考了国际上成熟的医疗器械法规,特别是美国FDA对医疗器械的审批要求,这也让全世界达成一致的医疗器械规范更进一步。
“严格的法规对整个医疗行业健康发展的影响总体上是积极的。”体外诊断上游医疗器械研发企业汇先医疗CEO颜菁认可道,“尽管欧盟MDR新规的实施将给中国出口企业带来成本增加、认证周期拉长和合规风险增加等问题,但法规对促进医疗器械厂商以更严格的标准要求自己,保障产品和标准的符合性,这对患者而言都是有利无害的,对整个医疗器械行业既是挑战,也是机遇。”
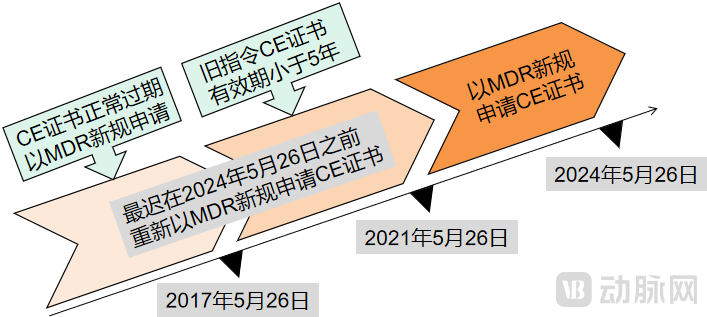
不同时期拿到CE证书企业的注意事项(动脉网制图)
对于在过渡期内依据旧指令MDD、AIMDD或IVDD公告机构签发的CE证书将在有效期内继续有效,但是从其交付日期起有效期不得超过5年,并且最迟于2024年5月26日失效。
而对于在过渡期之前获得的CE证书已经上市的医疗器械产品,需要在旧CE证书过期/失效之前(同样最迟于2021年5月26日),尽快重新申请基于MDR新规的CE证书:确认其产品风险分级是否升级,以及确认原CE证书的发证机构是否还具备MDR授权公告的资格,尽快修改原CE技术文件,再次向具有MDR发证资质的NB机构提出新的认证申请。
这也就意味着2024年之后,整个欧盟市场将不再存在基于旧法规的CE证书,医疗器械研发商尽快适应新规是当务之急,“提早做好迎接新规准备的医疗器械研发商在未来收入将会增加,而那些欧盟本土20%-30%的中小医疗器械厂商将会面临出局的局面。”3D打印医疗器械公司智塑健康CEO张靖表示。
与此同时,由于欧盟申请门槛的提高,以及对医疗器械研发过程的监管,国内传统通过OEM代工生产再到欧盟进行产品CE认证的路径也将彻底行不通,以代工生产医疗器械为核心业务的企业也将被迫转型。但这对于整个医疗器械欧盟市场的发展无疑是重大利好的,欧盟是仅次于美国的全球第二大市场医疗器械市场,占比全球医疗器械市场总和的26%,所以维护医疗器械企业的市场准入至关重要。
















